
William Gibson: A general strategy for TP53 missense mutant cancers with prototype small molecules
Harold J. Burstein, Breast cancer specialist and professor at Dana Farber Cancer Institute, shared a post by William Gibson, Medical Oncologist at Dana-Farber Cancer Institute, on X, adding:
“This is very cool stuff.”
Quoting William Gibson’s post:
“TP53:
-Discovered 45 years ago, most cited gene all time
-No therapies
-500 million people currently living will die of TP53 mutant cancers without new therapies
Our Preprint:
– A general strategy for TP53 missense mutant cancers (majority) with prototype small molecules.
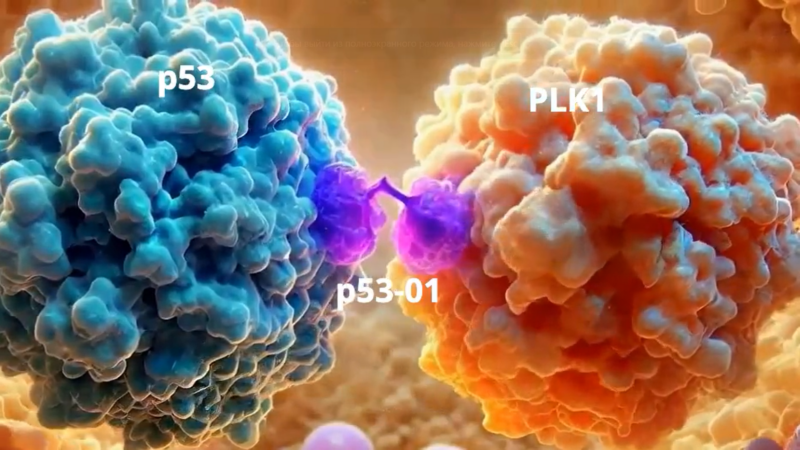
p53 protein abundance is a therapeutic window across TP53 mutant cancers and is targetable with proximity inducing small molecules
Autors: , , , , ,

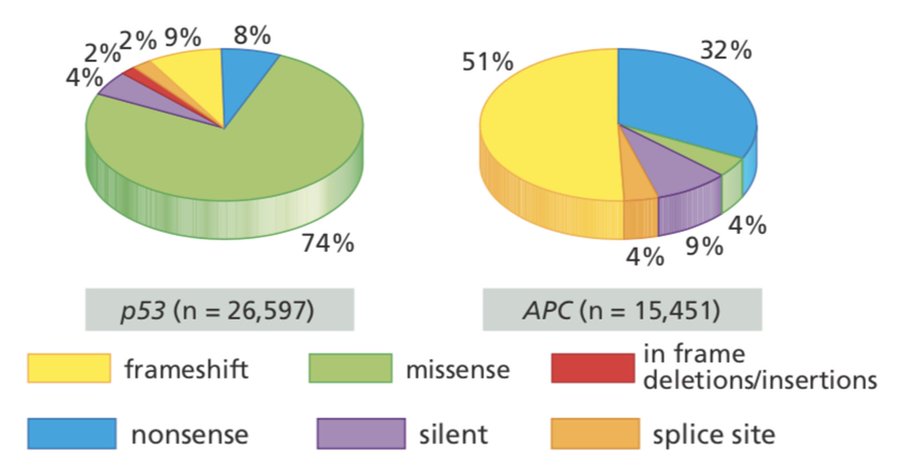
Some brief context: p53 is the most important tumor suppressor protein in humans. In contrast to most tumor suppressor genes, which are inactivated by nonsense or frameshift mutations, most TP53 mutations are missense mutations.
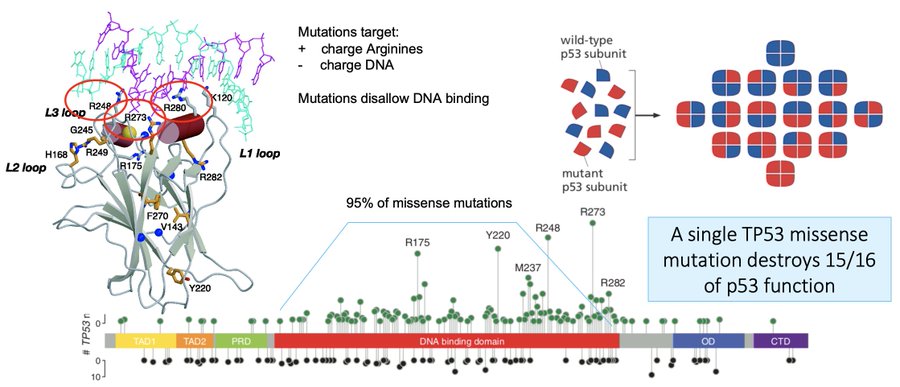
95% of these missense mutations occur in the DNA binding domain, and stop a whole p53 tetramer from being able to transcribe target genes. Because they poison the tetramer, a single missense mutation destroys 15/16th of p53 function; evolution prefers this path towards cancer.
In the past, we’ve hoped that synthetic lethals might allow us a way to selectively kill TP53 mutant cancers. Unfortunately, CRISPR screens now show us that synthetic lethals for p53 loss do not exist. Nature never owed us that favor.
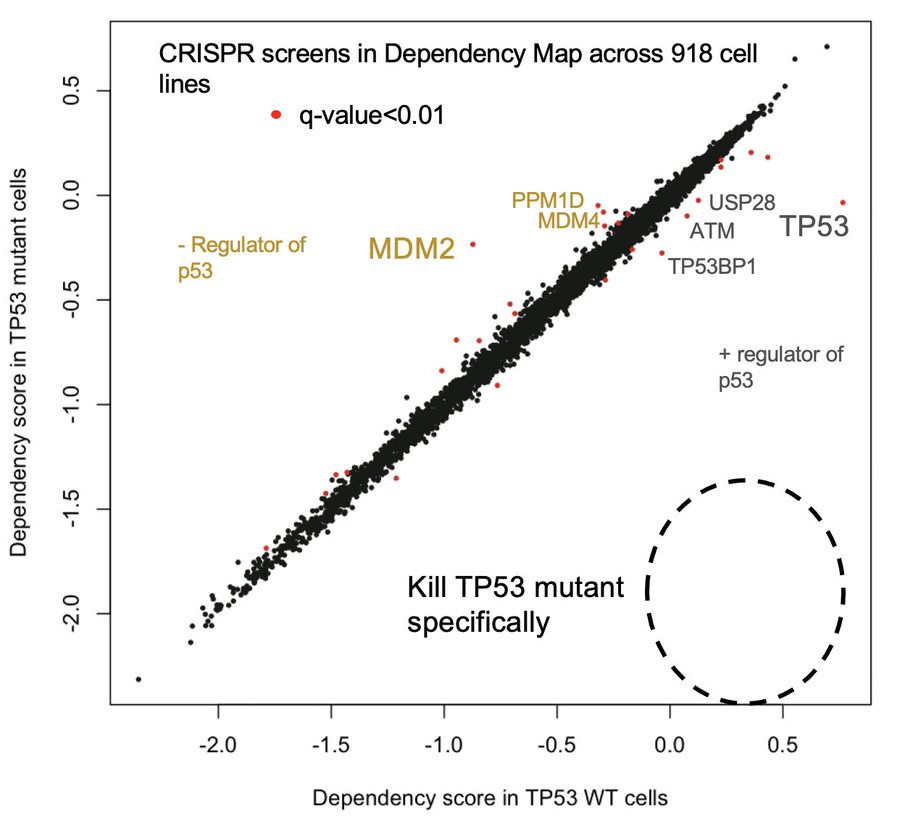
So is there anything we can use to distinguish TP53 mutant cancer cells from healthy wild-type cells?
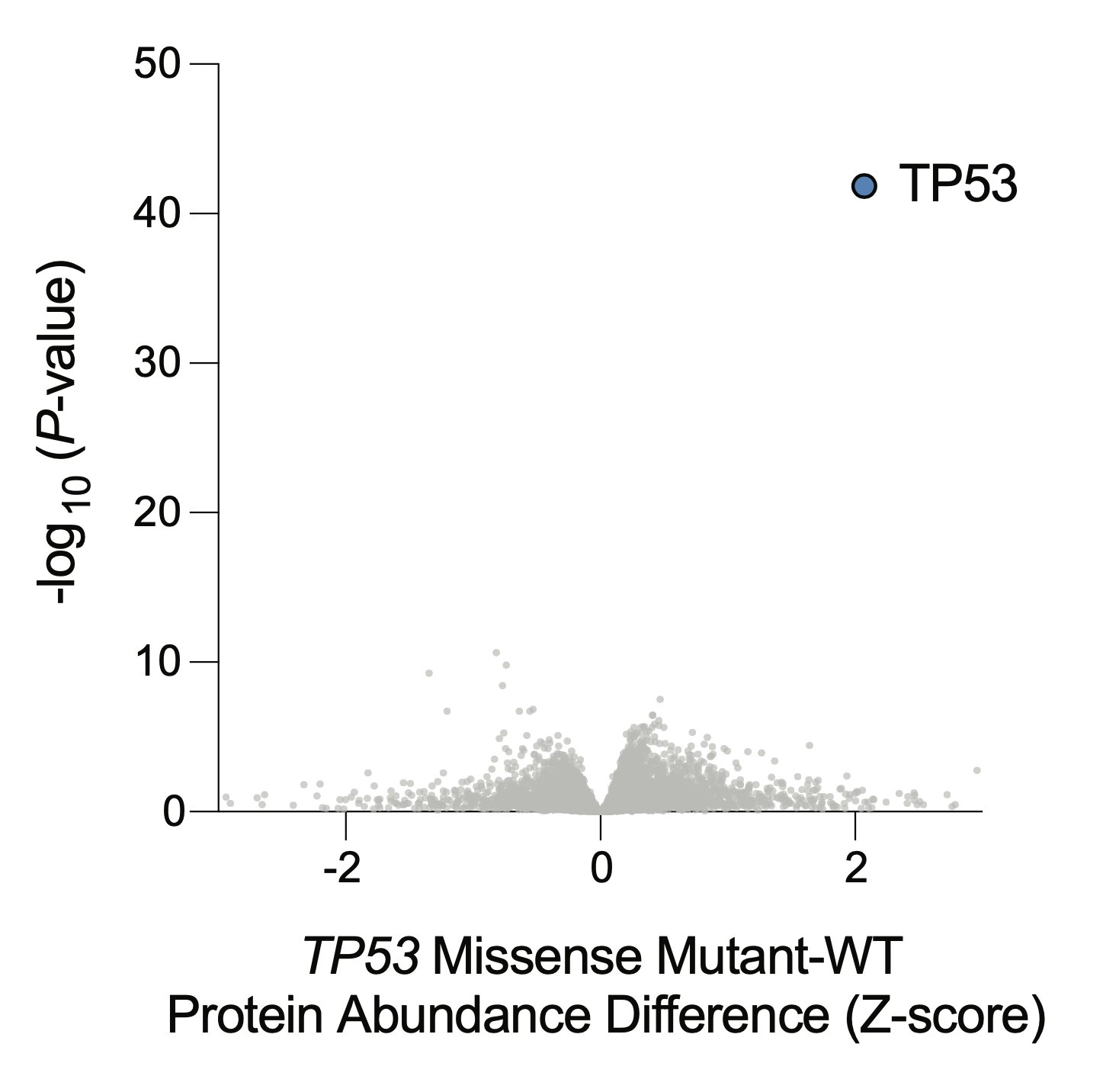
Only one protein is differentially abundant between TP53 mutant cells and WT cells: p53 itself. Normally, cells keep p53 at nearly undetectable levels, but it accumulates massively in cancer cells with missense mutations as they try to activate p53 signaling, but can’t.
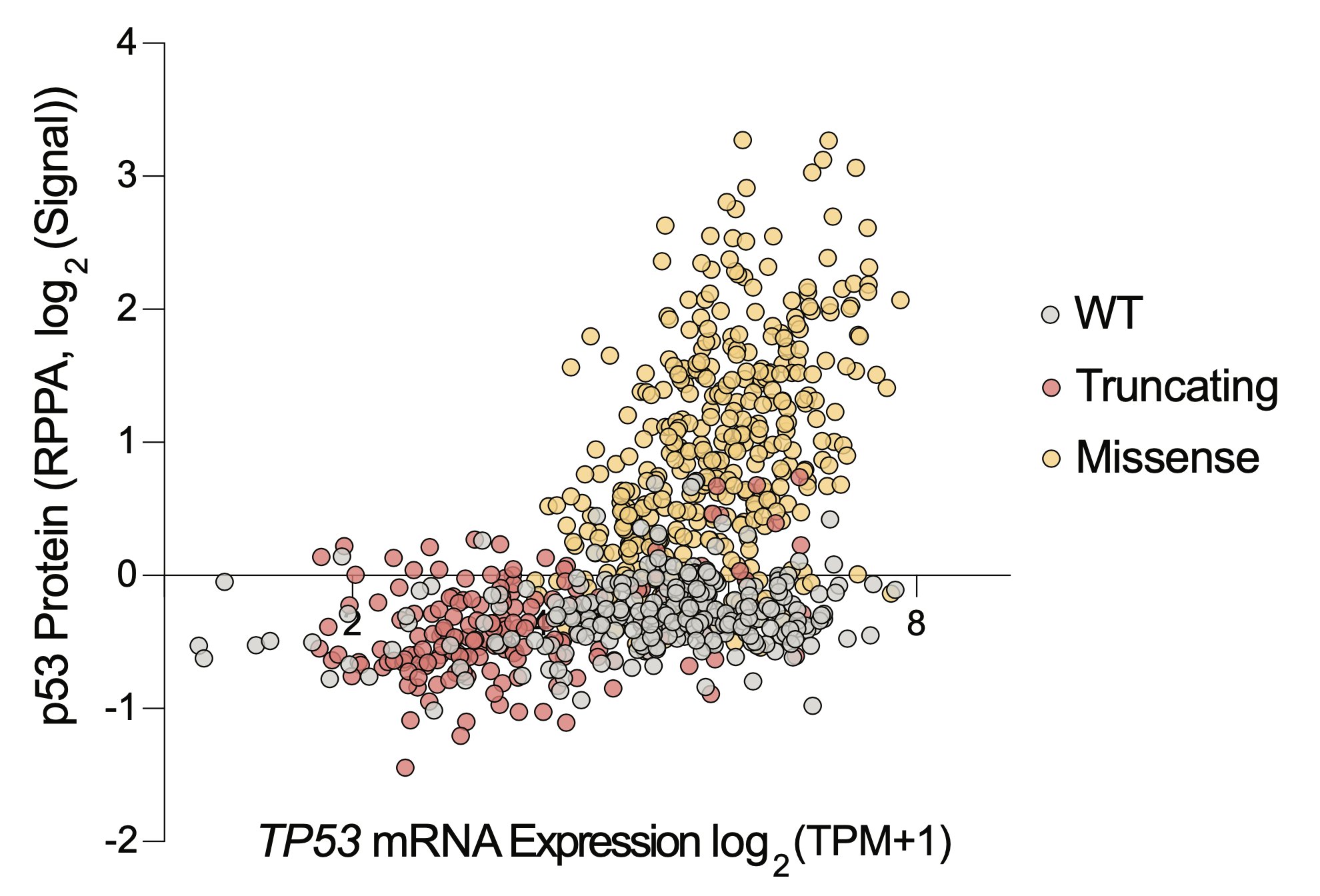
Thus, to make a selective therapy, you never needed to get mutant p53 to do what it was doing before (ie transcribe target genes), you just needed to make the accumulated p53 kill cancer cells. So we sought to make a drug that killed cells in proportion to p53 protein abundance.
While I had thought that I would need to find a p53-dependent molecular glue, a recent paper from Kanak Raina and Craig Crews showed that even simple bifunctional compounds against an accumulated intracellular protein can be used to induce selective lethality in cancer cells.
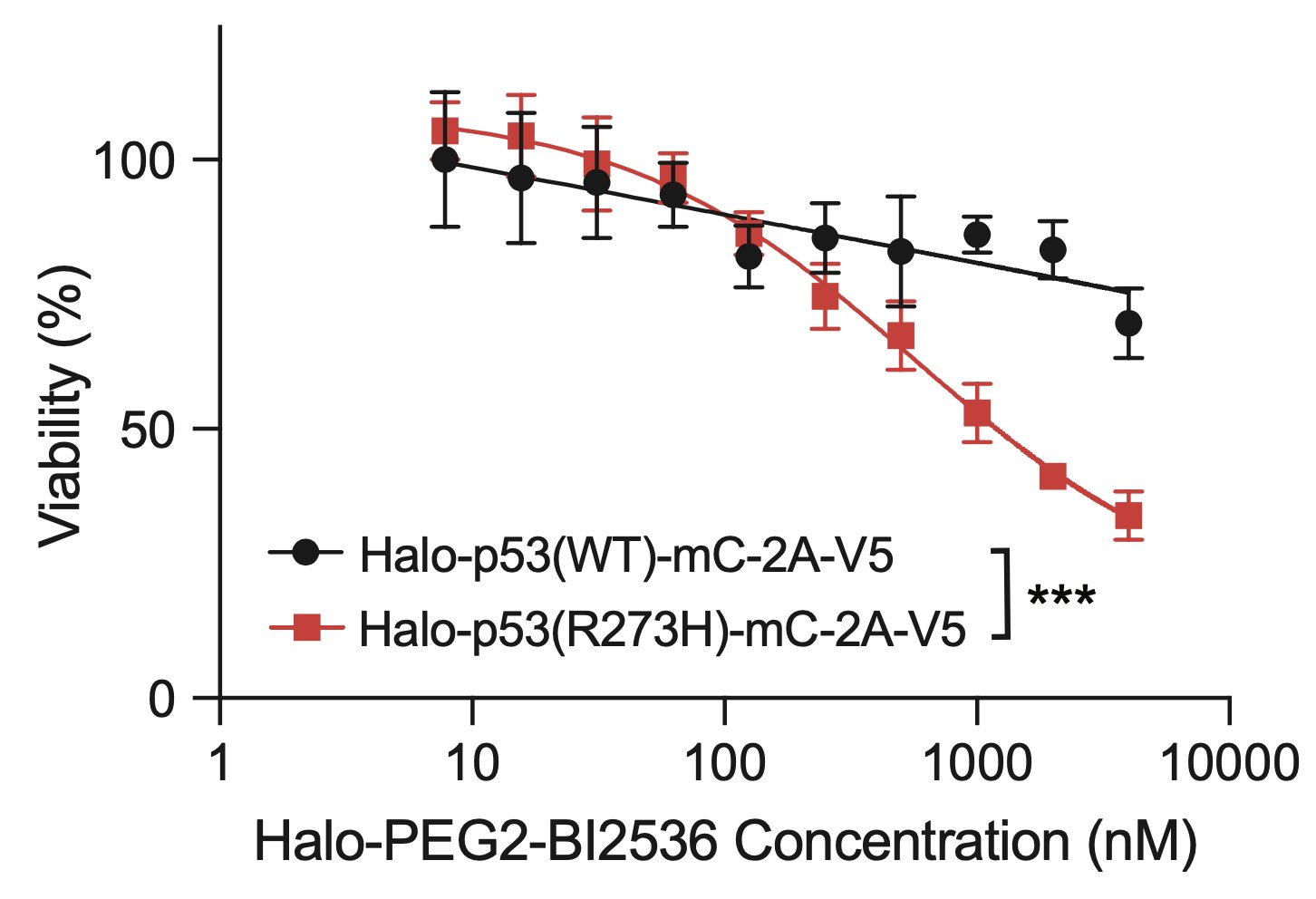
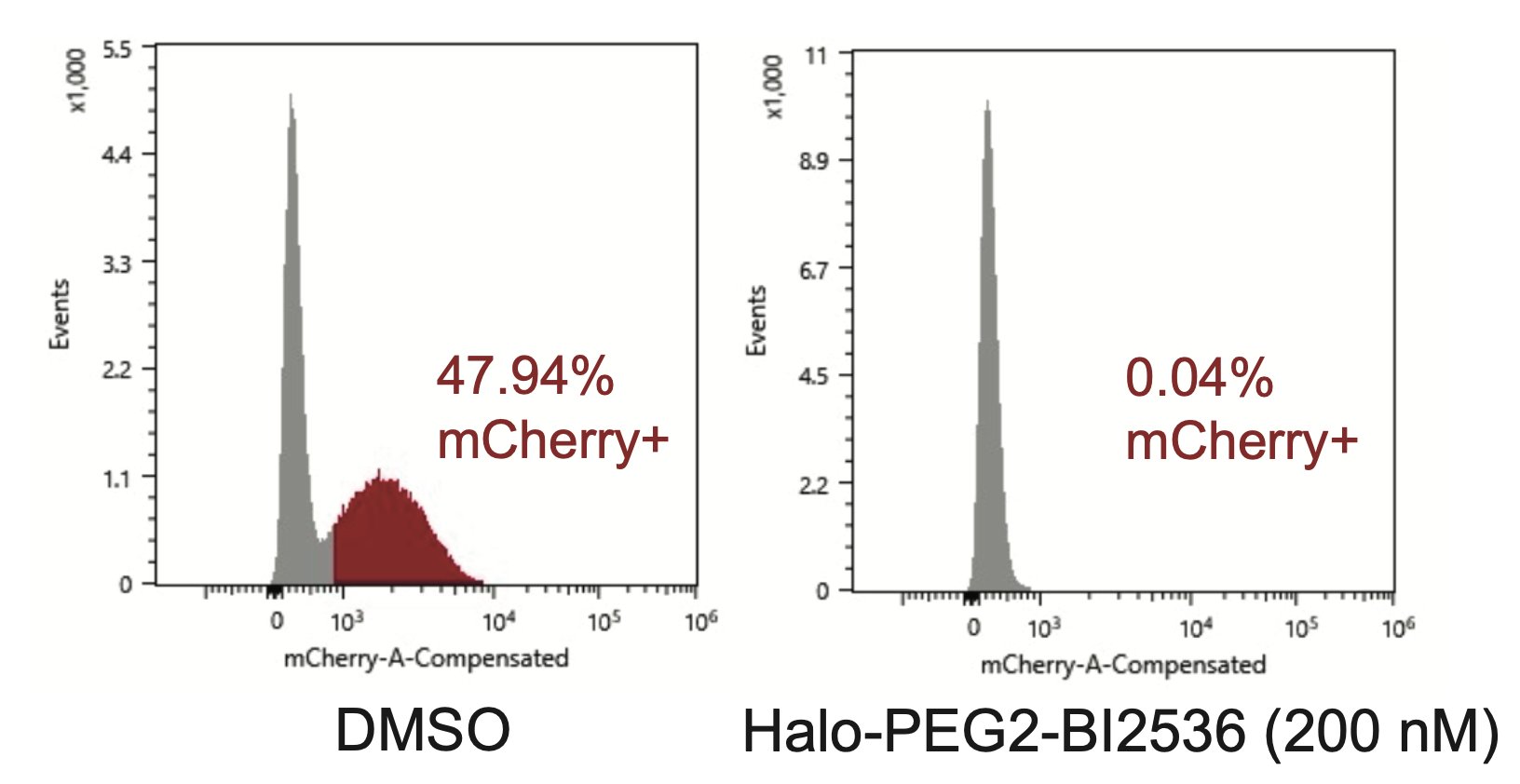
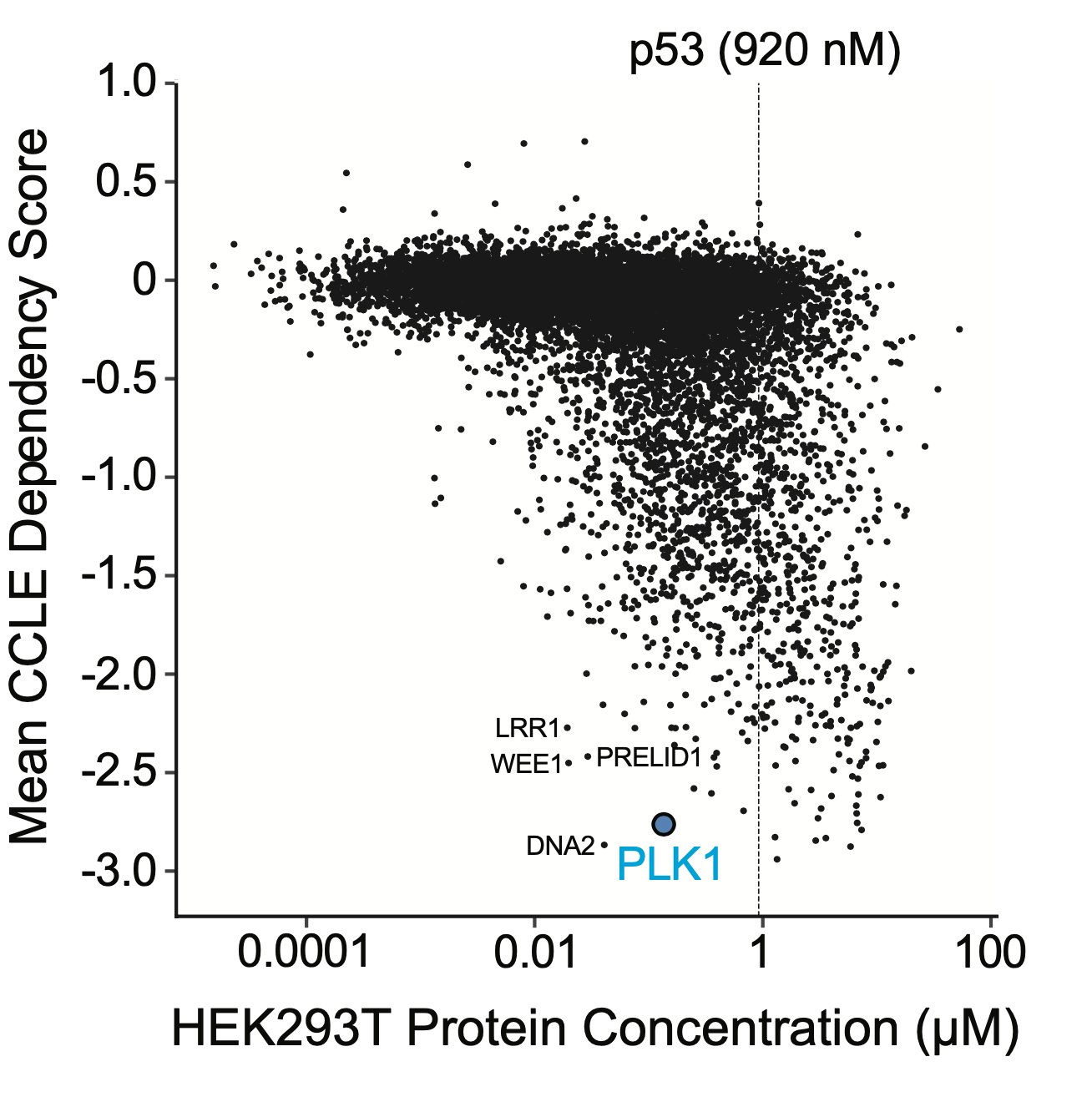
To pursue this strategy, we nominated PLK1 and WEE1 as a highly essential genes that are low in abundance, with high-quality inhibitors. We synthesized proof-of-concept bifunctional compounds using a Halo tag system (high quality WT p53 small molecule probes do not exist).
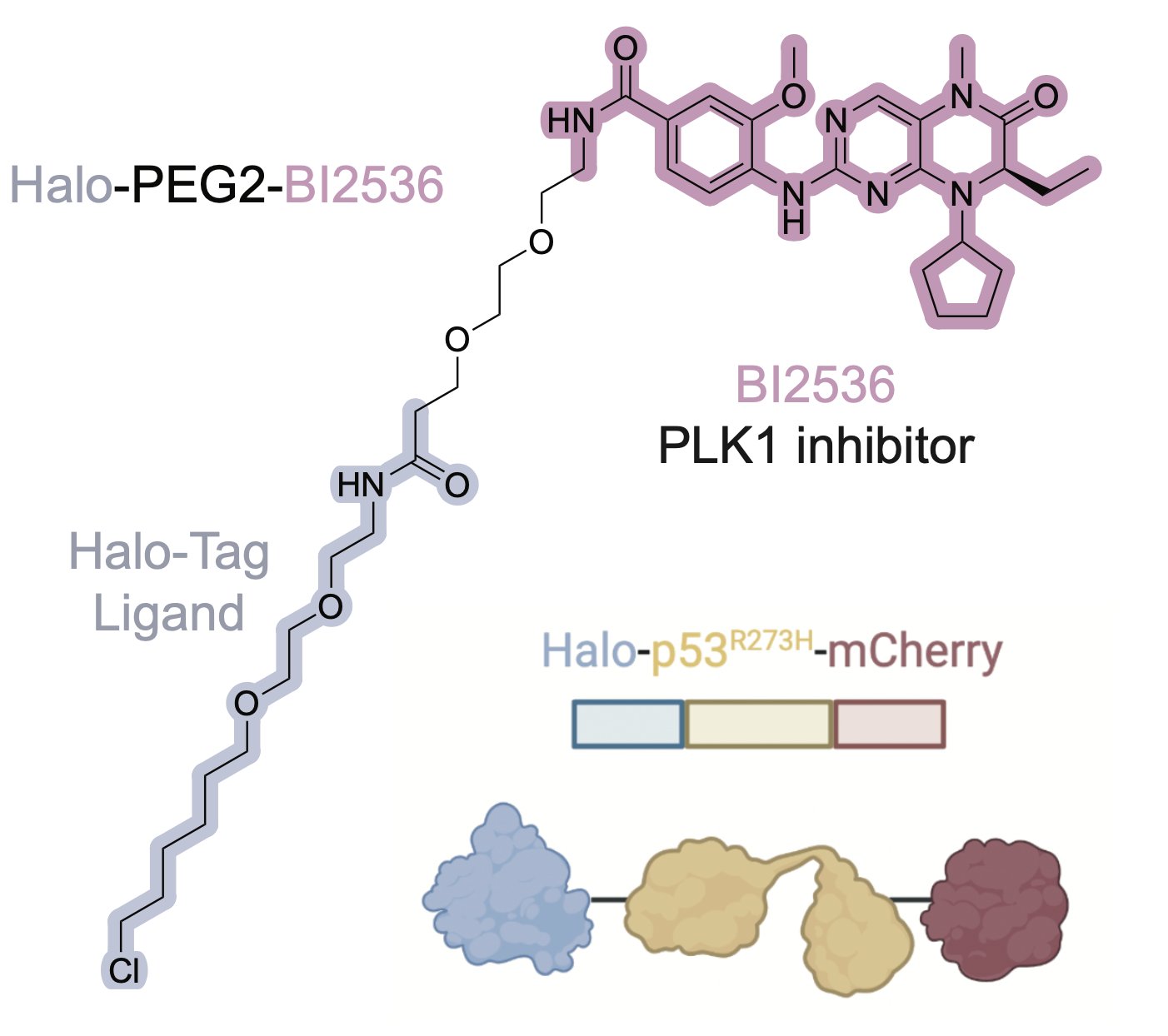
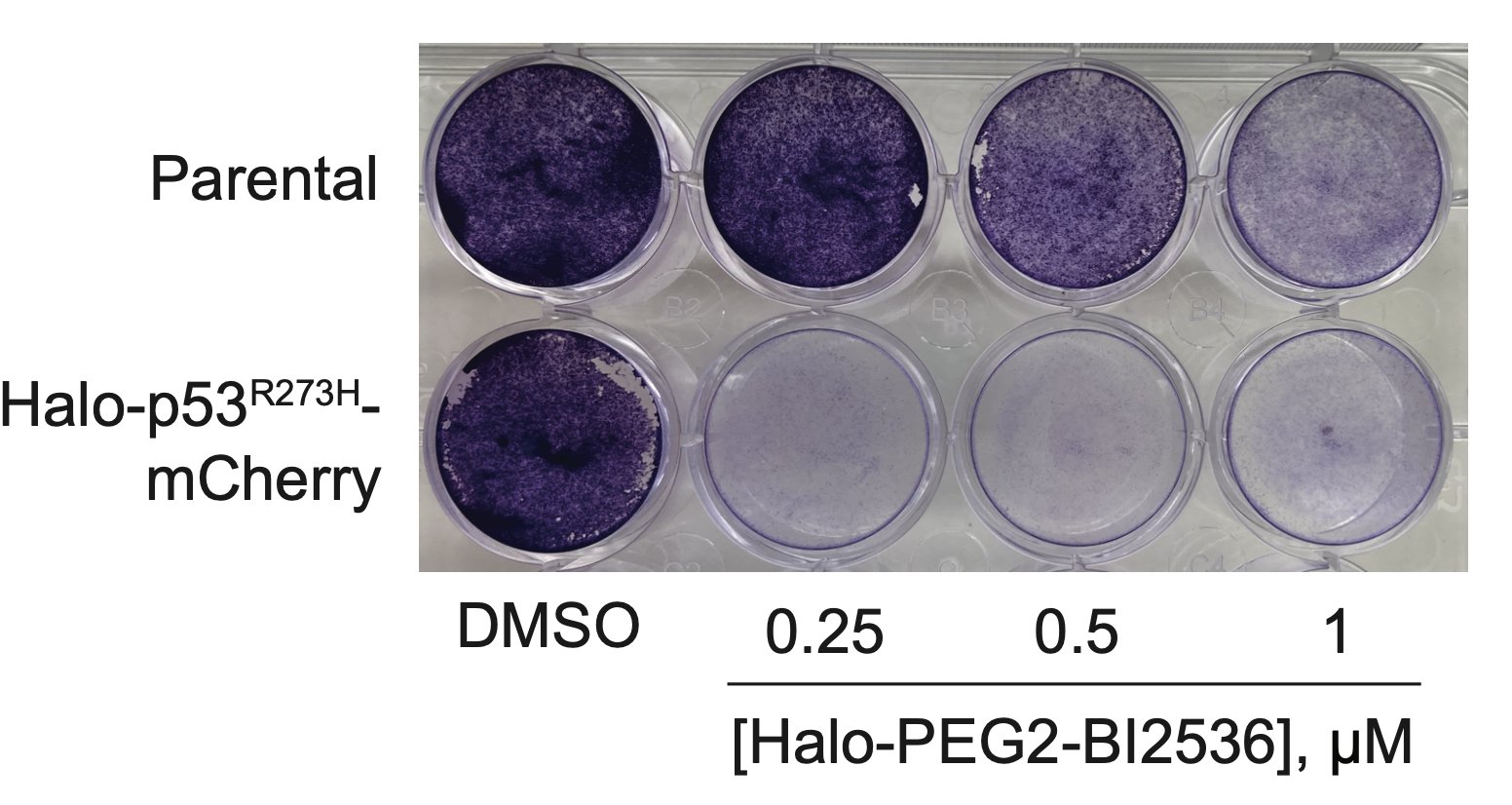
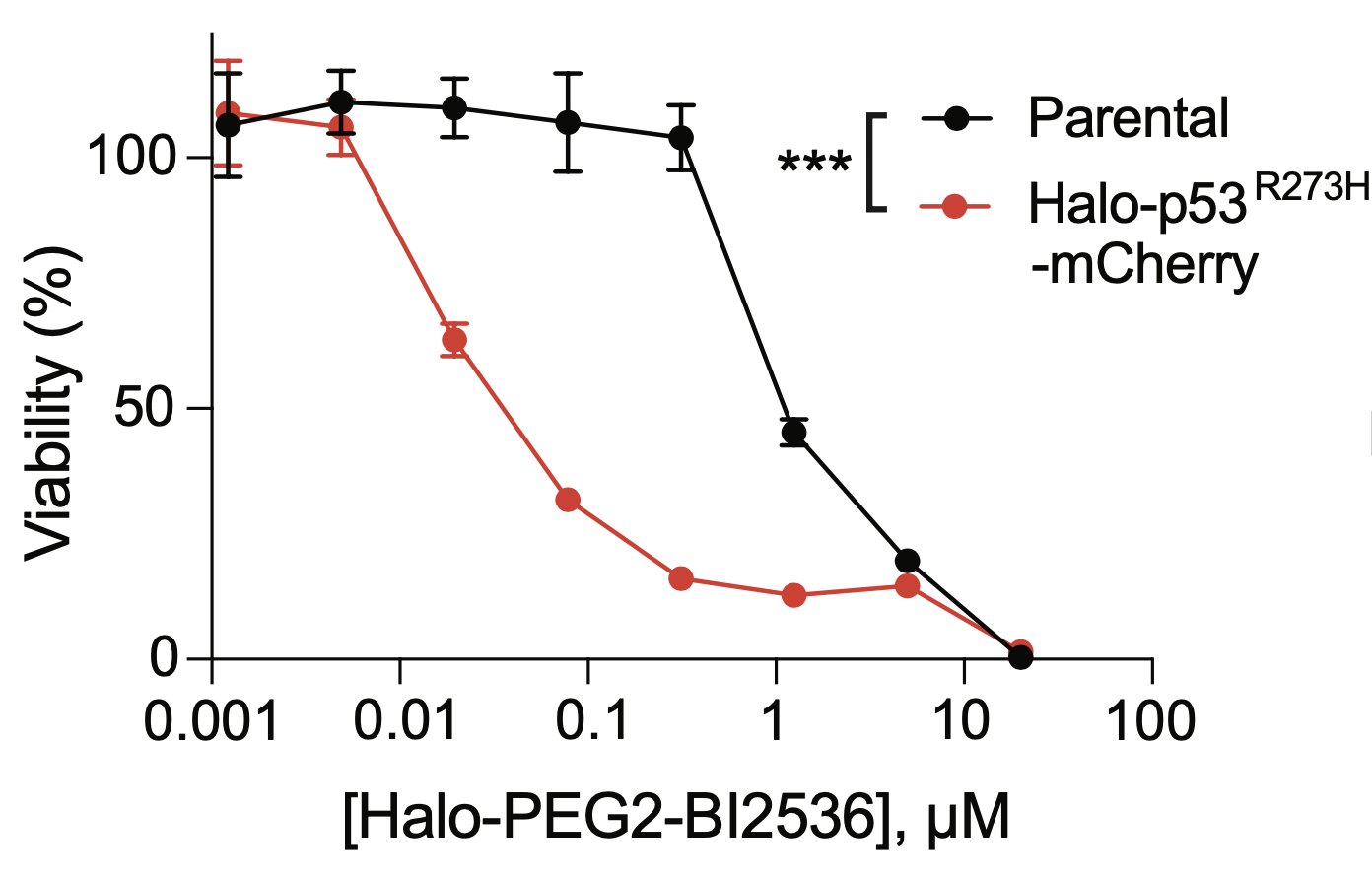
The bifunctional compound worked! It showed enhanced toxicity in cells expressing Halo-p53(R273H)-fusion proteins in several viability and competition assays.
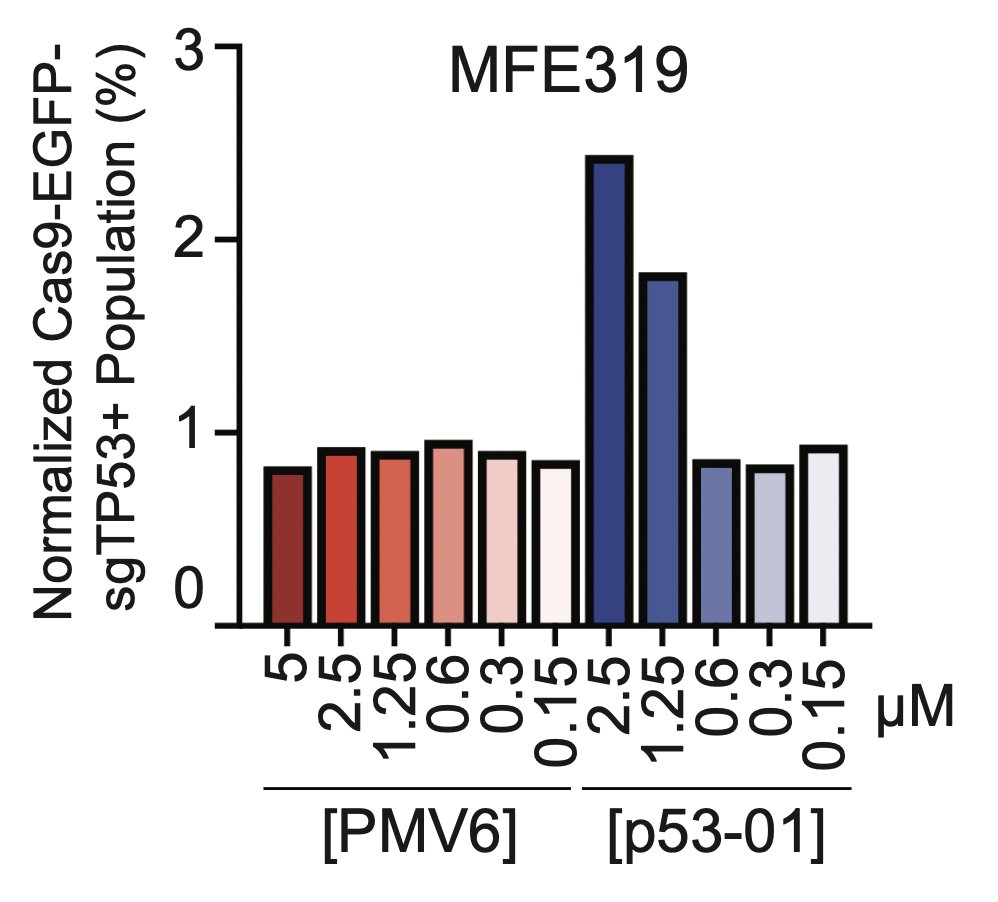
We wanted to bring this therapeutic model closer to a native application. We surveyed the patent literature and identified a p53Y220C binding pharmacophore, typified by the depicted compound 6 from a PMVP Pharma patent. We made an analogous bifunctional, calling it p53-01.
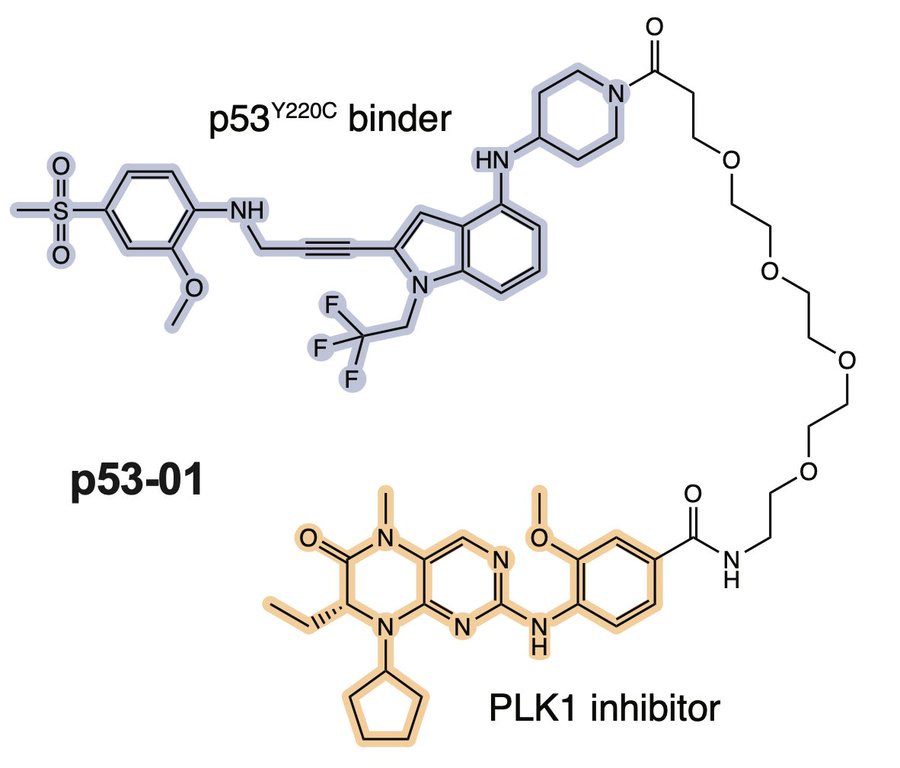
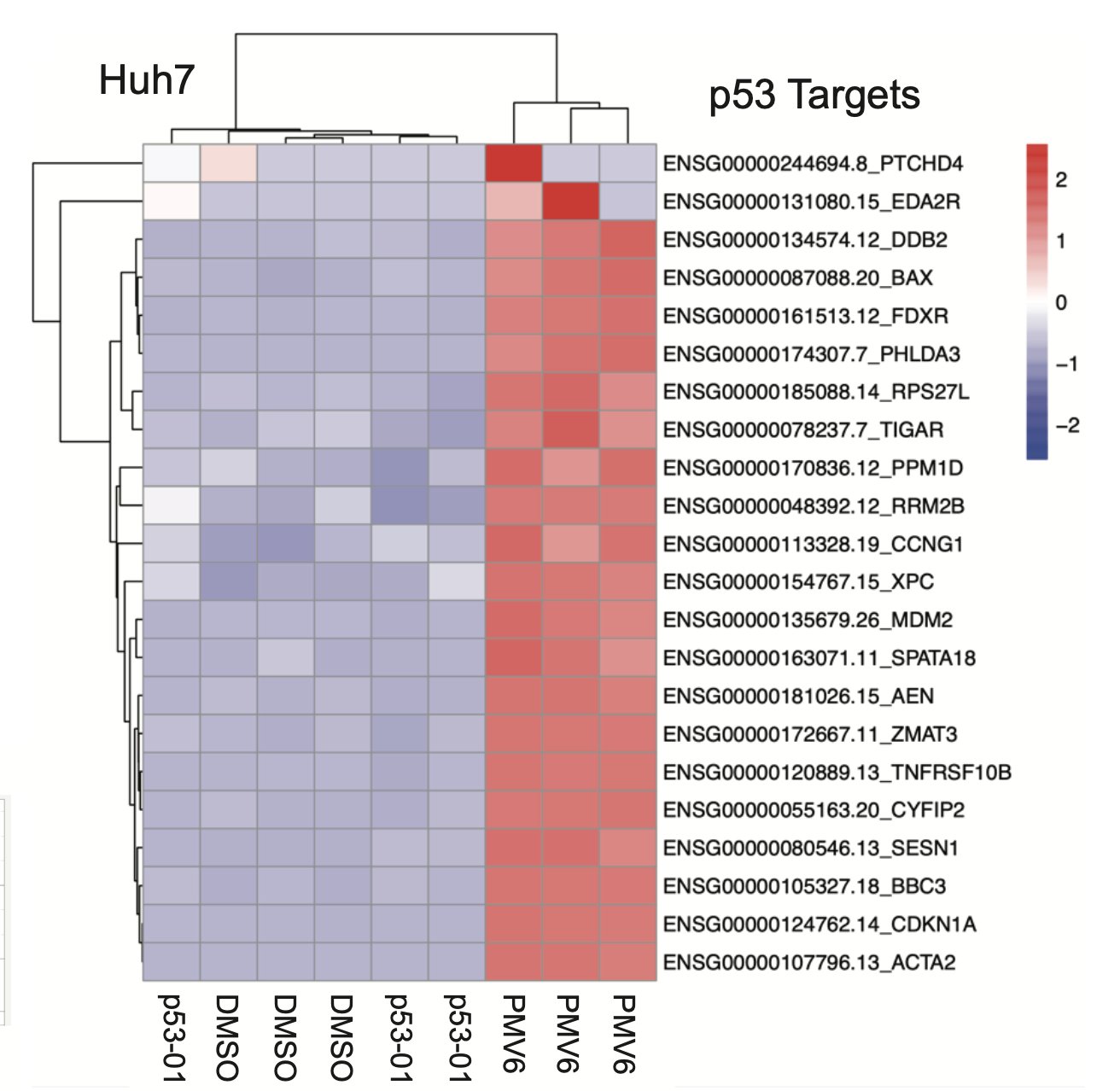
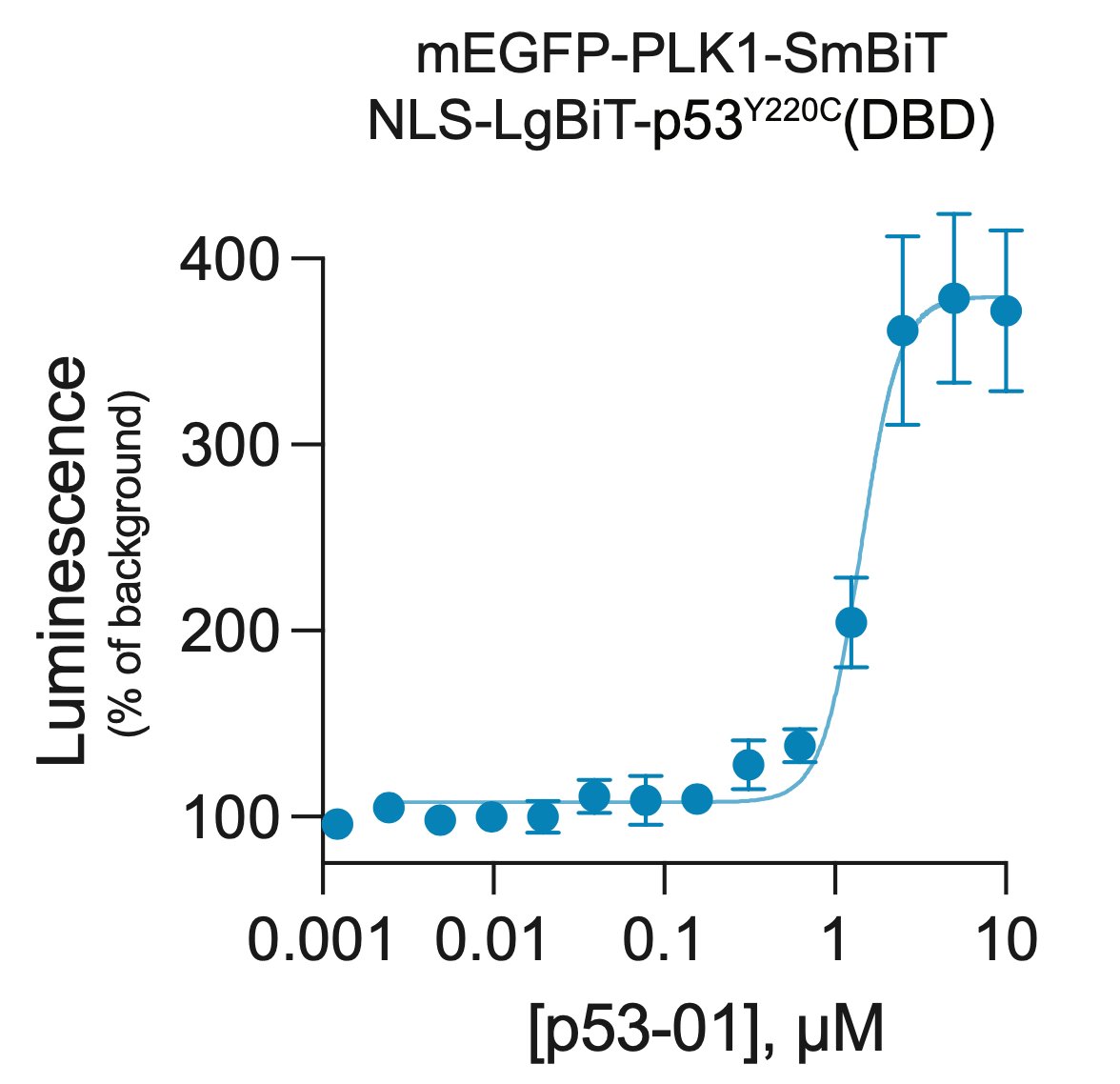
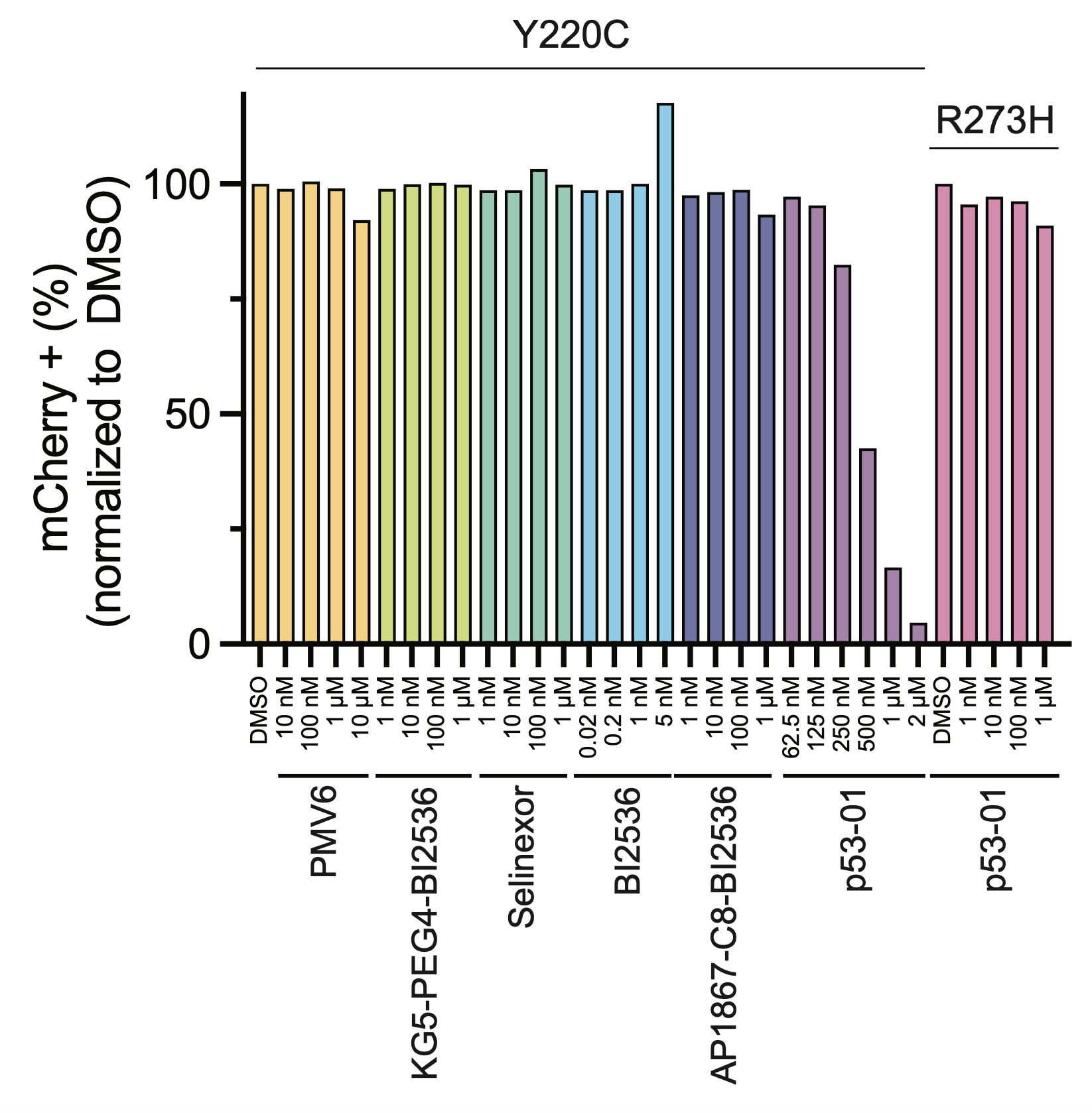
p53-01 worked! It caused ternary complex formation, and enhanced toxicity in p53(Y220C) expressing cells, where the monovalent binders had no effect. Importantly, the mechanism was not p53-refolding -> transcription, as the transactivation domain of p53(Y220C) was deleted.
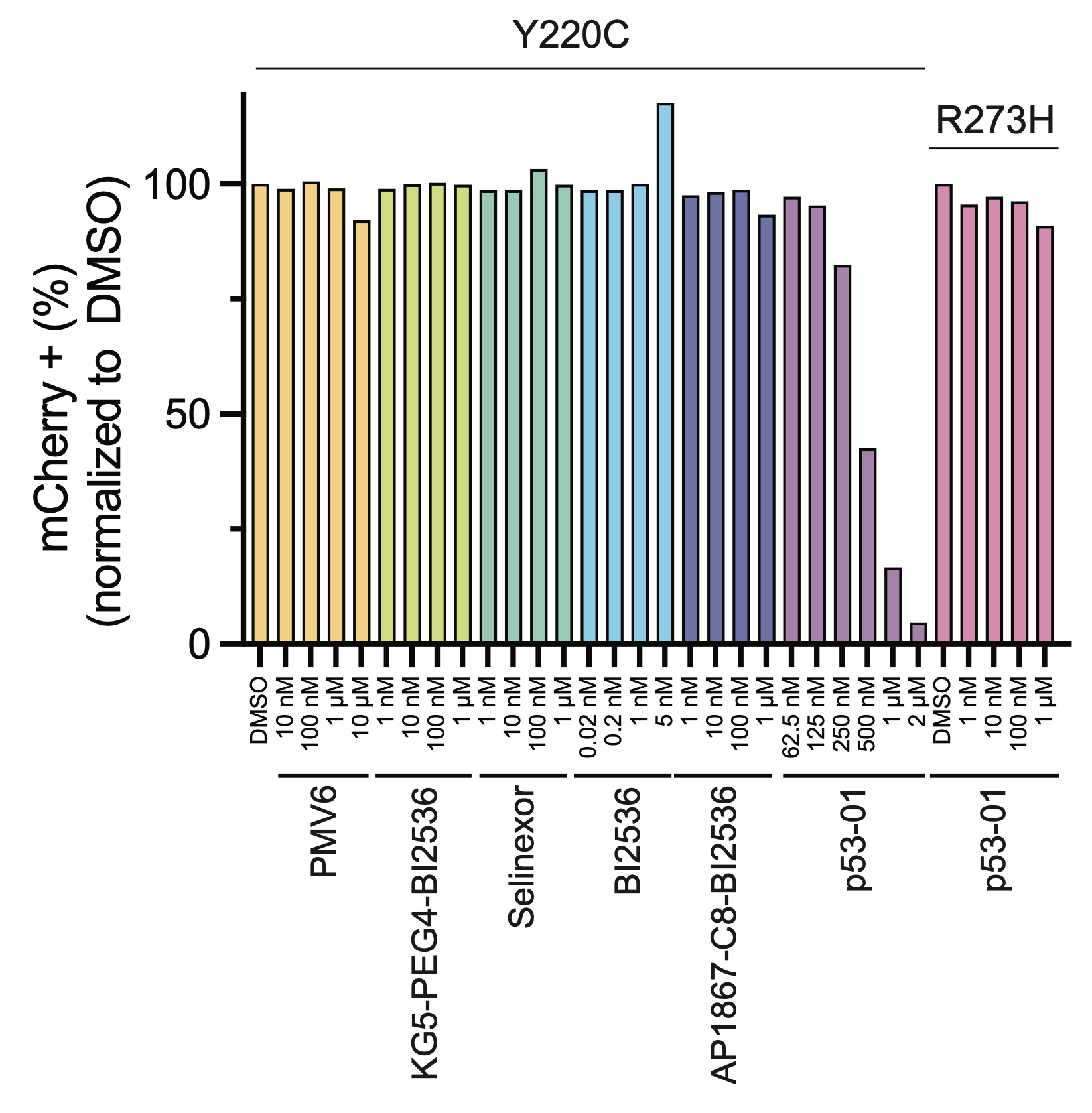
The enhanced toxicity could be rescued by competing off p53(Y220C) binding by addition of the monovalent binder, confirming a p53(Y220C)-dependent mechanism of action.

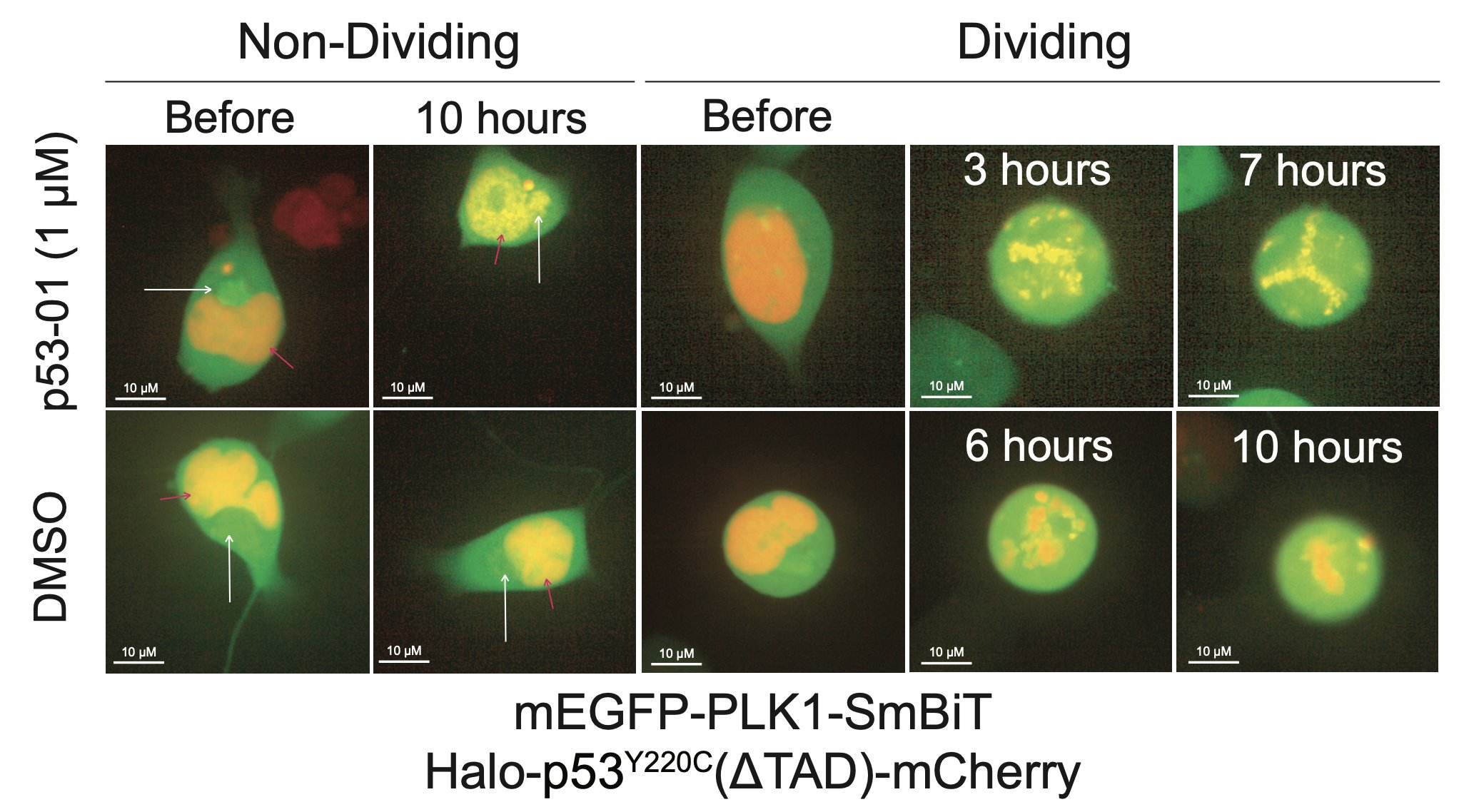
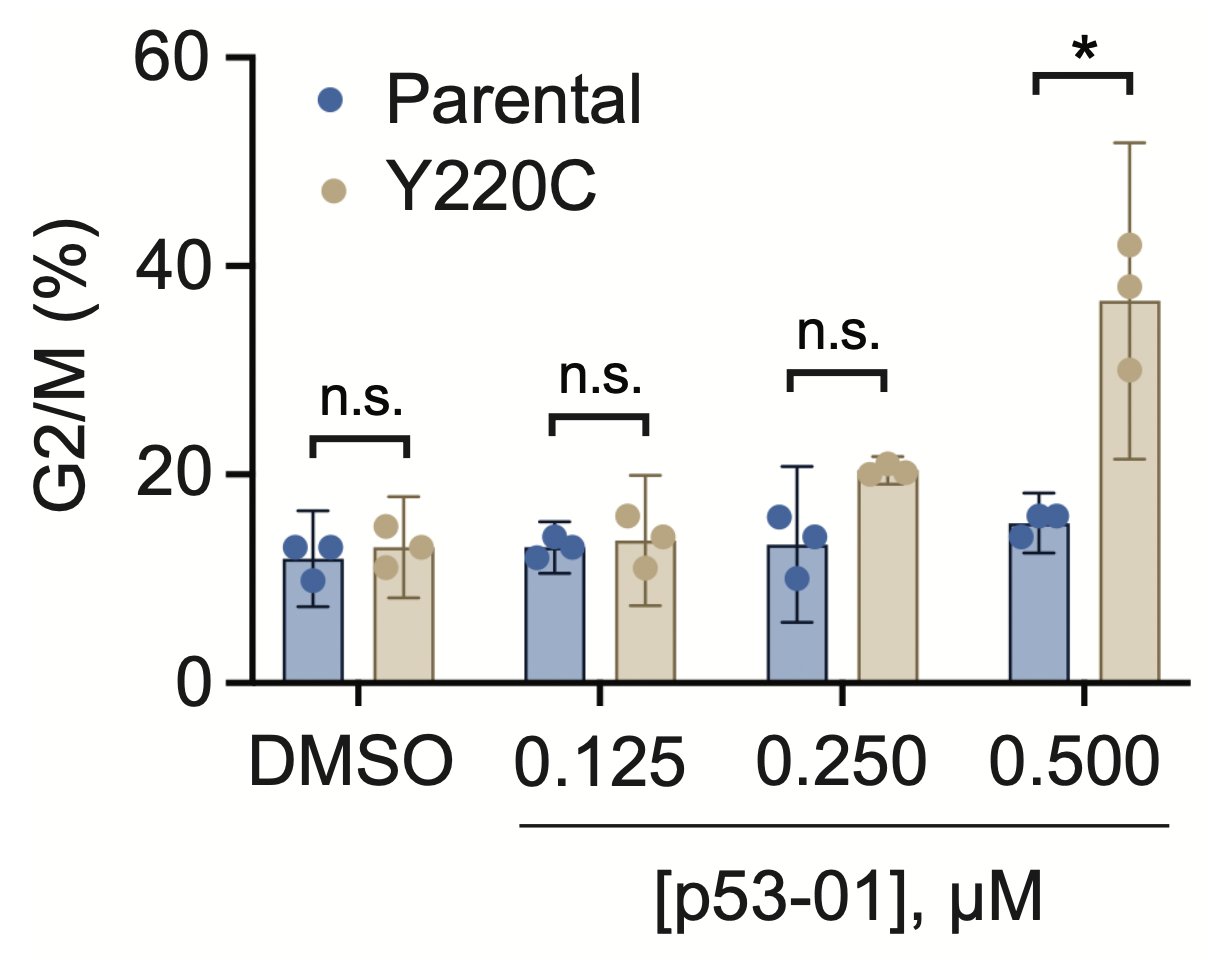
p53-01 caused co-localization of PLK1 and p53(Y220C), driving PLK1 out of the centrosome and inducing G2/M arrest.
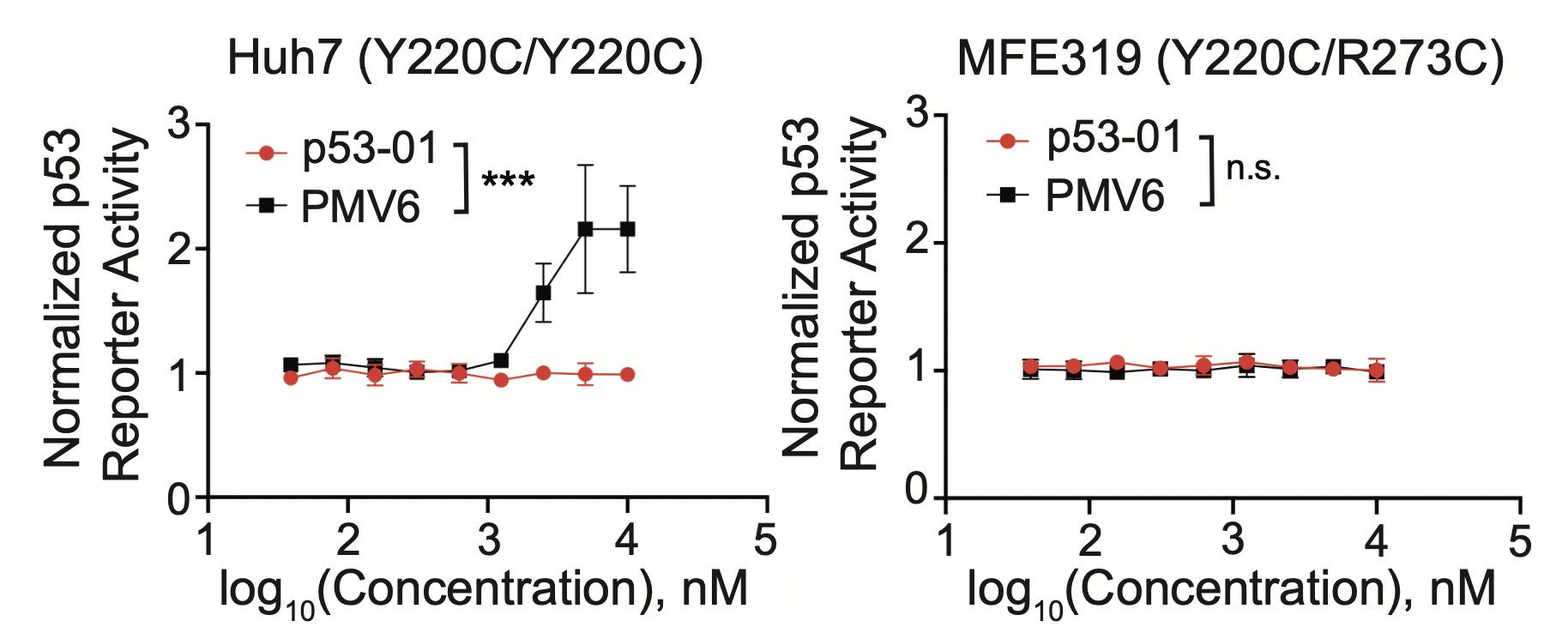
In contrast to the monovalent binder, p53-01 does not work by turning on p53 activity. Instead, it has a gain-of-function mechanism (enhanced PLK1 inhibition) that allows it to work in an endogenous context in cancer cell lines with other TP53 hotspot mutations in trans.
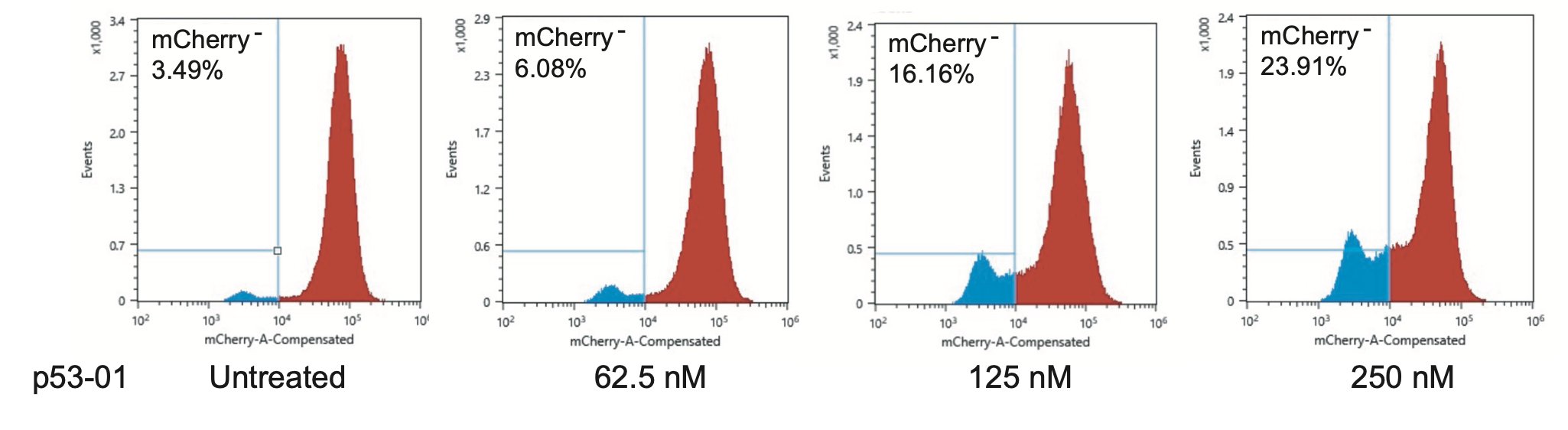
We confirmed that the mechanism of p53-01 was p53-dependent in cell lines with endogenous TP53(Y220C) mutations using CRISPR/Cas9 gene editing. We also observed that cells cultured in p53-01 for several weeks, lost p53(Y220C) expression to gain resistance to the compound.
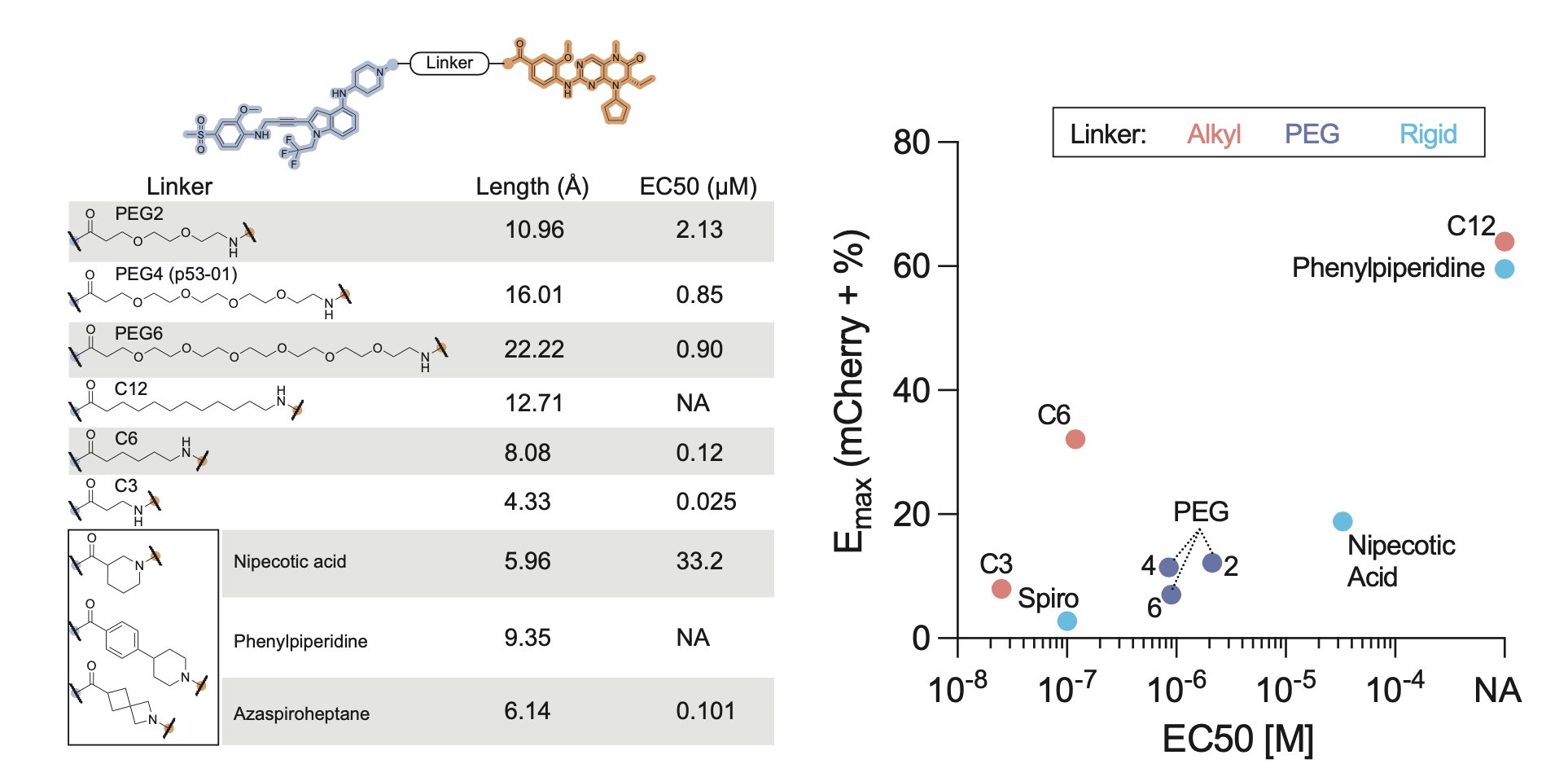
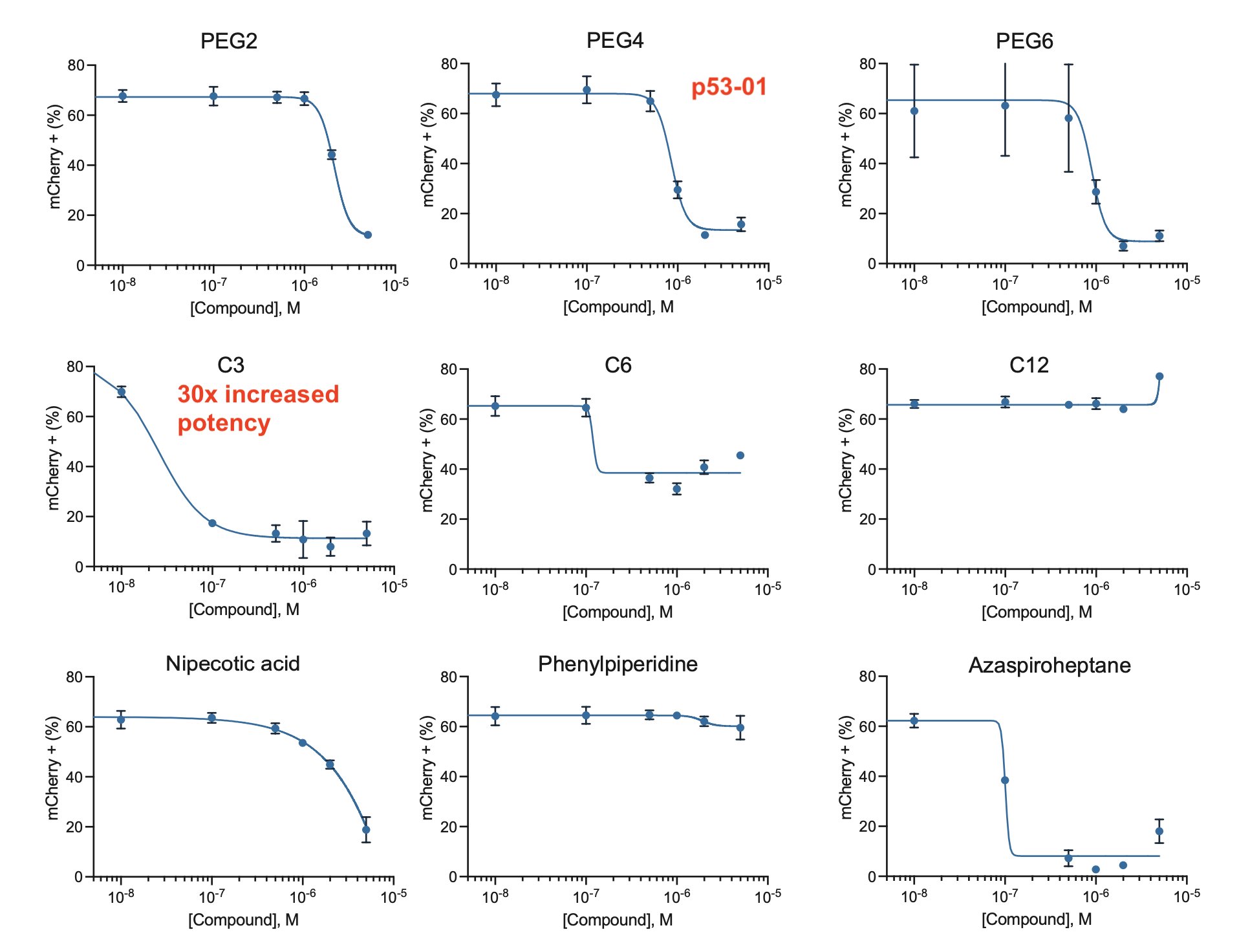
Finally, we made a small series of analogs.
Some had no activity, but one showed a 30-fold increase in potency compared to p53-01!
Shorter linkers seemed to work best.
I want to emphasize: these compounds are so, so early and so far from helping people. They’re clunky, they might have terrible metabolic/PK/PD properties..
But I think we do see a way forward. In the fullness of time we might expect highly potent, cooperative, gain-of-function molecular glues that finally “drug” TP53 and drive important biologic responses in the millions of patients that desperately need new options.
We’ll be working to expand this work to pan-p53 targeting compounds, and to make these early prototypes into more effective, glue-like compounds.
Many thanks to my enormously supportive recent/former* mentors, Stuart Schreiber and Matthew Meyerson. *Speaking of, new announcement…
And it’s been an absolute pleasure working with the extraordinarily talented incoming Harvard PhD student Ananthan Sadagopan. He is a force of nature, and key to the success of this work.
Footnotes:
– The 500 million living people figure comes from Scott Lowe’s “Putting p53 in context” Cell review in 2017.
– I also think other molecular glues involving p53 will be possible! The natural world already made one by accident as Dan Nomura showed with asukamycin.”
Source: Harold J. Burstein/X and William Gibson/X
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}