Nicholas McGranahan: Welcome to the CONIPHER TREEtorial, just in time for Christmas
Nicholas McGranahan, Group leader at the CRUK-UCL Lung Cancer Centre of Excellence, shared on X/Twitter:
“CONIPHER, our method for automated reconstruction of tumour subclonal structure and phylogeny that we used in our recently published TRACERx studies, is now out in Nature Protocols! (See article here).
Welcome to the CONIPHER TREEtorial, just in time for Christmas!
Tumours are heterogeneous mixtures of distinct tumour clones as a result of a complex evolutionary process. Reconstructing tumour phylogenies from many samples, affected by complex structural alterations, and with variable levels of noise remains difficult.
To address these TREEmendous challenges and help in analysing the large TRACERx cohort we developed CONIPHER, a tool to automate tumour phylogenetic reconstruction while dealing with various sources of noise and errors, and scaling to large numbers of samples and mutations.
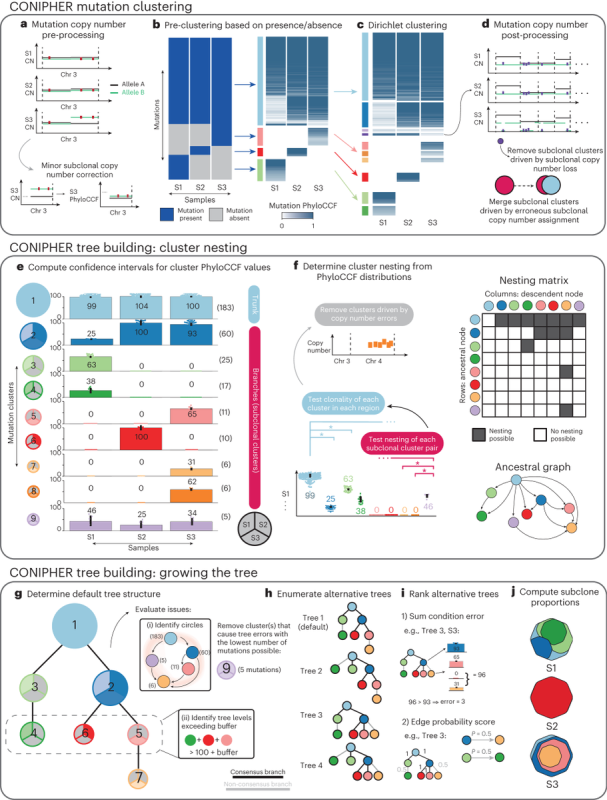
Stage 1: CONIPHER aims to cluster mutations to identify tumour subclones, which reflect the different branches of the tumour’s phylogeny. Critically, CONIPHER takes into account that the frequency of a mutation may reflect its copy number state.
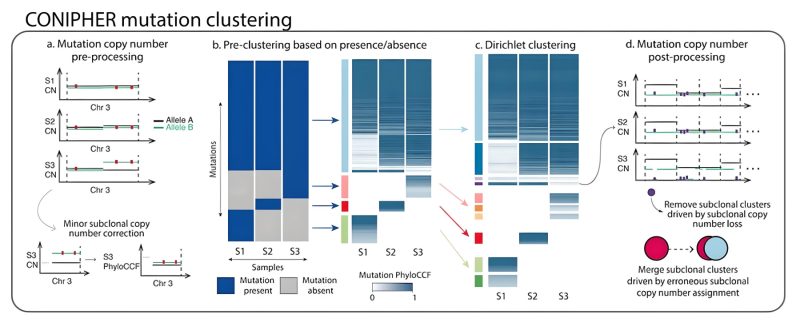
Stage 2: CONIPHER enumerates the all evolutionary plausible ancestral/descendent relationships between subclones. In this stage, false subclones likely driven by missed copy number events are removed.
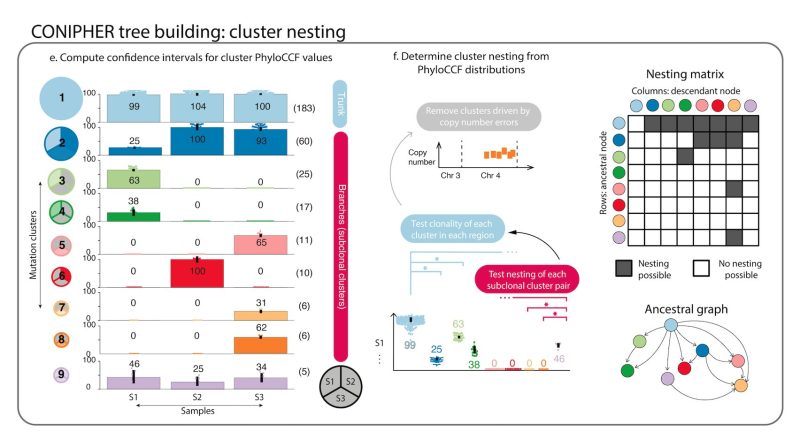
Stage 3: CONIPHER uses inferred evolutionary relationships between clones to create a tree. It removes erroneous clones leading to tree cycles and those breaking the pigeonhole principle and crossing rules, resulting in a phylogeny maximising no. mutations retained on the tree.
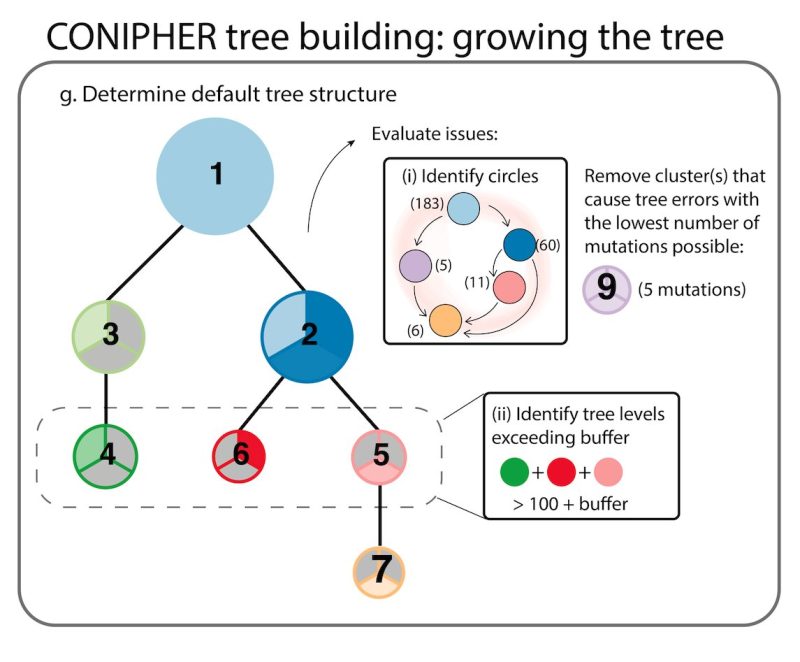
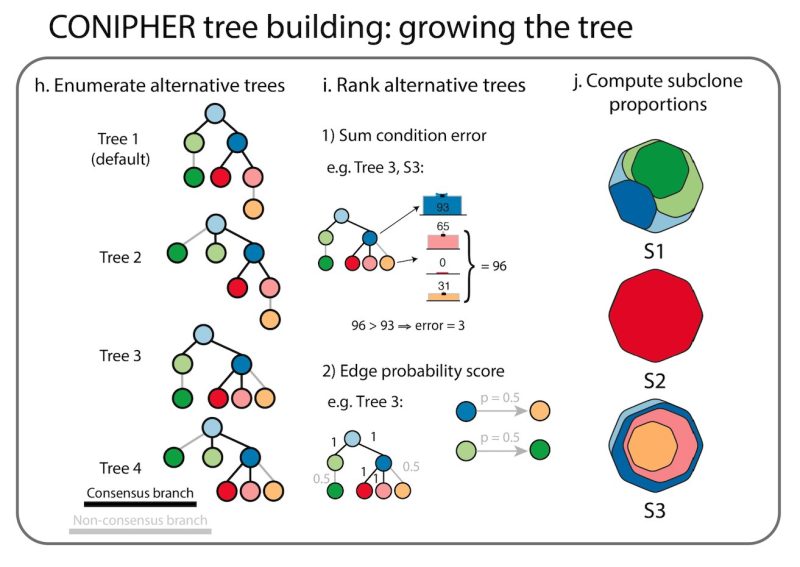
Stage 4: using the final subclones identified, CONIPHER can then enumerate all possible phylogenies, and rank which trees have the lowest error. CONIPHER also automatically outputs clone proportions in each sample.
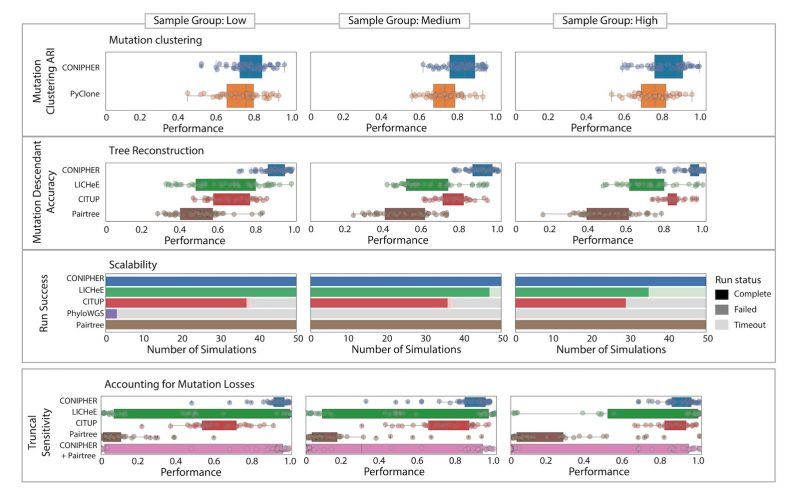
Based on simulations, we find CONIPHER performs well for a high number of samples, correctly classifies truncal and subclonal mutations and correctly identifies erroneous clusters to remove.
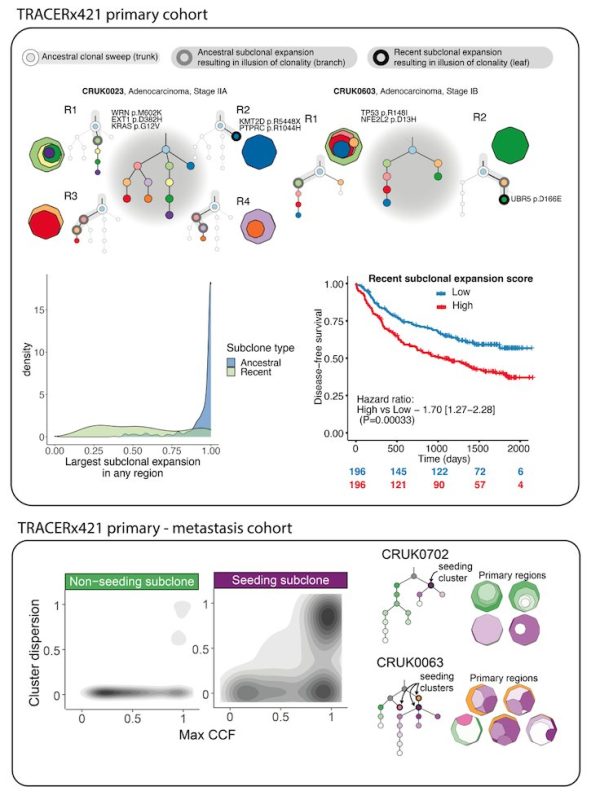
We used CONIPHER to reconstruct phylogenies for the full TRACERx421 primary and paired primary-metastasis cohort . A full wiki for exploring the data (using either R or Python) is publicly available here.
CONIPHER enabled us to identify expanded subclones dominating an entire sample. Recent subclonal expansions were found to be associated with poor outcome in primary tumours, and subclones seeding metastasis were found to dominate several regions of the primary tumour.
CONIPHER is a user-friendly package and can be installed through conda (see here). An example wrapper script to run from command line (see also). See the protocol for a full description of the method and how to run it!
We would like to thank the patients and families, and our funders, without whom this research would not be possible Rosetrees Trust, Wellcome, Science and Innovation at Cancer Research UK, Cancer Research UK, CRUK Lung Centre, CRUKCOLcentre, UCL Cancer Institute, The Crick.
Finally, thanks to co-authors Kristiana Grigoriadis, Ariana Huebner, Abi Bunkum, Emma Colliver, Alex Frankell for working on the method and manuscript and Charles Swanton, Simone Zaccaria for co-supervising. We hope that CONIPHER can be a useful tool for the community!”
For more details click here.
Source: Nicholas McGranahan/X